Forbidden and allowed: what symmetry does to a spectrum
Dissolve cobalt(II) chloride in water and you get a solution so pale it barely counts as pink. Add concentrated hydrochloric acid and it flips to a saturated royal blue. The ion is the same Co²⁺ throughout, the transitions are the same family of \(d\)–\(d\) excitations, and the color change from pink toward blue is the familiar story of a shifting energy gap. But the intensity change is not: the octahedral \([\mathrm{Co(H_2O)_6}]^{2+}\) absorbs with a molar absorptivity of order 1–10 M⁻¹cm⁻¹, while tetrahedral \([\mathrm{CoCl_4}]^{2-}\) comes in around a hundred times stronger1. Nothing about the energy gap explains a factor of a hundred. What changed is that the water complex has a center of symmetry and the chloride complex does not.
The pigments post in this series leaned on exactly this fact — its Figure 3 drew site symmetry as a dial running from forbidden (pale octahedral viridian) through partly allowed (tetrahedral cobalt blue) to fully allowed (the trigonal-bipyramidal site of YInMn blue) — and then left a promissory note: what “symmetry,” “forbidden,” and “allowed” mean is the subject of a later post. This is that post. The claim to be earned is that hue and strength are set by two independent dials — the gap sets the color, the symmetry sets the intensity — and the machinery that earns it is one integral, one parity argument, and one small table (Figure 1).
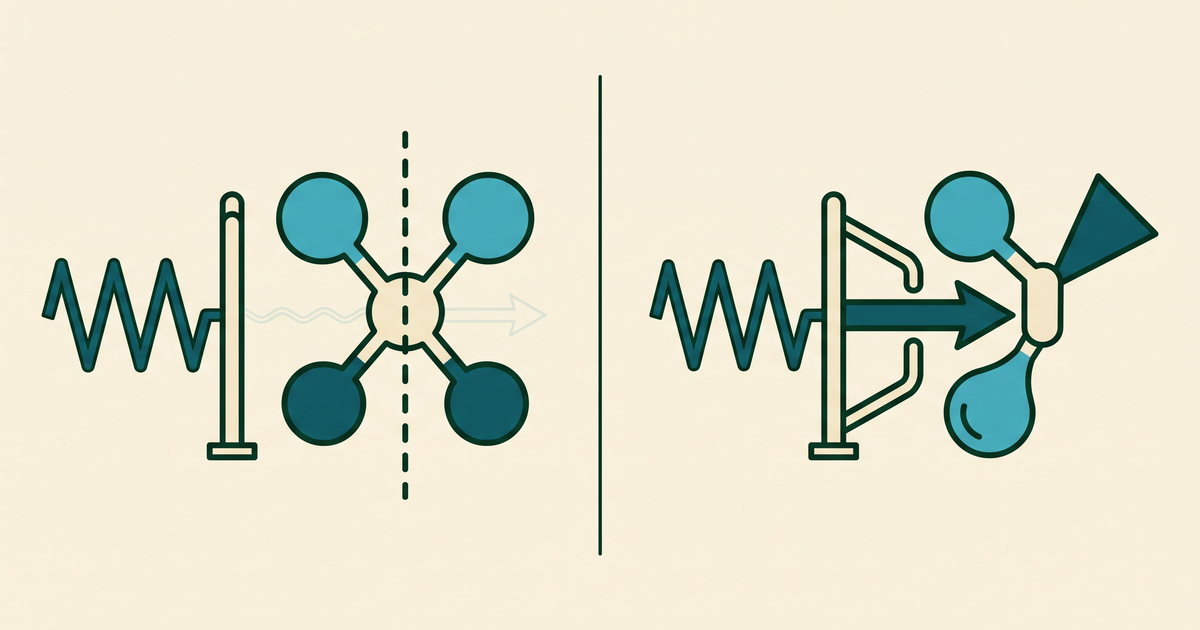
Figure 1. Symmetry as a gate. Two molecules with the same energy gap face the same light wave; the centrosymmetric one absorbs it feebly, the asymmetric one strongly. The gap decides which photon fits; the symmetry decides how hard the molecule can grab it.
The integral that decides
Everything reduces to one quantity this series has already met. The matrix-element post traced molar absorptivity, refractive index, and the Pockels effect back to the transition dipole moment,
\[ \boldsymbol{\mu}_{fi} \;=\; \langle \psi_f \,|\, \hat{\boldsymbol{\mu}} \,|\, \psi_i \rangle \;=\; \int \psi_f^{*}\, \hat{\boldsymbol{\mu}}\; \psi_i \, d\tau , \]
and the absorptivity post packaged its square into the dimensionless oscillator strength \(f\) — of order 1 for the strongest transitions, and proportional to the band area you actually measure. The intensity of any absorption band is, up to constants, \(|\boldsymbol{\mu}_{fi}|^2\).
A selection rule is nothing more mysterious than a statement about when that integral is exactly zero for reasons of symmetry alone — before you know anything about the detailed shapes of \(\psi_i\) and \(\psi_f\), their energies, or the strength of the light. If symmetry forces \(\boldsymbol{\mu}_{fi} = 0\), the transition is forbidden: the state exists, the photon energy can match the gap perfectly, and still — to first order — nothing happens. If symmetry permits \(\boldsymbol{\mu}_{fi} \neq 0\), the transition is allowed, which is a license, not a guarantee: how large the integral actually is remains a question about wavefunctions, the kind the push–pull post answered with TD-DFT. Symmetry only ever gives one of two answers — exactly zero or no comment — and that binary verdict is worth several orders of magnitude in a spectrum.
An odd function integrates to zero
The entire argument, in its simplest form, is a fact from first-year calculus: the integral of an odd function over a symmetric interval vanishes, because every positive contribution on the right is cancelled by a mirror-image negative contribution on the left (Figure 2). All the group theory below is this one cancellation, generalized.
Figure 2. The whole machinery in one picture. Left: an odd integrand — every positive patch of area has a mirror-image negative twin, and the total is exactly zero no matter what the detailed shape is. Right: an even integrand — the mirror halves add instead of cancelling. A selection rule is the statement that symmetry makes the integrand of \(\langle \psi_f | \hat{\mu} | \psi_i \rangle\) odd.
Now apply it to the transition dipole. In any molecule or crystal site that possesses a center of inversion — a point through which every atom maps onto an identical atom — every electronic state can be classified by parity: gerade (\(g\), even, unchanged by inversion) or ungerade (\(u\), odd, sign-flipped). The dipole operator \(\hat{\boldsymbol{\mu}} = -e\hat{\mathbf{r}}\) is built from the coordinates themselves, and coordinates flip sign under inversion: \(\hat{\boldsymbol{\mu}}\) is odd, always and everywhere. So the integrand of the transition dipole between two \(g\) states is \(g \times u \times g = u\) — odd — and the integral vanishes. Same for \(u \rightarrow u\). Only \(g \leftrightarrow u\) survives:
\[ g \rightarrow g \;\; \text{forbidden}, \qquad u \rightarrow u \;\; \text{forbidden}, \qquad g \leftrightarrow u \;\; \text{allowed}. \]
That is the Laporte rule, and it is the entire explanation of the pale octahedral pigments. The five \(d\) orbitals are all even functions — \(l = 2\), parity \((-1)^l = +1\), gerade — so in a site with an inversion center every \(d\)–\(d\) transition is \(g \rightarrow g\): forbidden. The chromium ions in viridian sit in octahedra; octahedra have inversion centers; therefore viridian is muted, however well its gap is tuned. The rule predates the machinery used here to derive it — Otto Laporte and William Meggers extracted it empirically from the term analysis of iron’s atomic spectrum in 1925, a year before the Schrödinger equation, as the observation that “even” terms combine only with “odd” ones2; Wigner later showed it is nothing but parity conservation. The atomic version is the familiar \(\Delta l = \pm 1\): \(s \rightarrow p\) allowed, \(d \rightarrow d\) and \(d \rightarrow s\) not.
Remove the inversion center and the argument evaporates — there is no parity label left to conserve. A tetrahedral site has no inversion center, so in \([\mathrm{CoCl_4}]^{2-}\) the \(d\)–\(d\) bands are no longer parity-forbidden (the metal \(p\) orbitals, odd, can mix into the same symmetry species as the \(d\)’s and lend them allowed character), and the hundredfold intensity jump of the opening paragraph follows. The trigonal-bipyramidal site that makes YInMn blue so intense is the same trick played deliberately: put Mn³⁺ where there is no center of symmetry and the transition is free to be strong3. The pigment post’s dial from forbidden to allowed is, literally, a dial of how thoroughly the site symmetry destroys the parity cancellation of Figure 2.
The character table: cancellation, bookkept
Parity handles molecules with an inversion center, but most molecules have lower, less tidy symmetry — a rotation axis here, a mirror plane there — and the cancellation argument still works; it just needs bookkeeping. That bookkeeping is group theory, and the working object is the character table4,5.
The water post already used this machinery without naming it: it sorted water’s orbitals into “labelled boxes” — \(a_1\), \(b_1\), \(b_2\) — under the point group \(C_{2v}\). Those labels are irreducible representations (irreps): the complete list of distinct ways any function attached to the molecule can behave under its symmetry operations. For \(C_{2v}\) — one two-fold axis, two mirror planes — the full table is small enough to show whole (Table 1), with the molecule in the \(yz\) plane as in the water post.
| \(C_{2v}\) | \(E\) | \(C_2\) | \(\sigma_v(xz)\) | \(\sigma_v'(yz)\) | linear function |
|---|---|---|---|---|---|
| \(A_1\) | \(+1\) | \(+1\) | \(+1\) | \(+1\) | \(z\) |
| \(A_2\) | \(+1\) | \(+1\) | \(-1\) | \(-1\) | — |
| \(B_1\) | \(+1\) | \(-1\) | \(+1\) | \(-1\) | \(x\) |
| \(B_2\) | \(+1\) | \(-1\) | \(-1\) | \(+1\) | \(y\) |
Table 1. The character table of \(C_{2v}\), the point group of water and formaldehyde. Each row is an irrep — a possible symmetry behavior — and each entry says what a function of that species does under the operation at the top of the column (\(+1\) unchanged, \(-1\) sign-flipped). The last column is the payload for spectroscopy: which component of the dipole operator transforms as which irrep. Note that no dipole component transforms as \(A_2\).
The generalization of “odd × odd = even” is that symmetry species multiply: the integrand \(\psi_f^* \,\hat{\mu}\, \psi_i\) belongs to the product of the three species, \(\Gamma_f \otimes \Gamma_\mu \otimes \Gamma_i\), computed by multiplying the \(\pm 1\)’s column by column. And the generalization of “an odd function integrates to zero” is:
The integral \(\langle \psi_f | \hat{\mu} | \psi_i \rangle\) can be nonzero only if \(\Gamma_f \otimes \Gamma_\mu \otimes \Gamma_i\) contains the totally symmetric irrep (the top row, all \(+1\)’s).
Anything that is not totally symmetric changes sign under some operation of the group — and then the same mirror-image cancellation of Figure 2 kills the integral, with that operation playing the role of the inversion.
Two worked examples, both from molecules this blog has already computed. First water: its HOMO is the out-of-plane lone pair \(1b_1\), its LUMO the \(4a_1\) antibonding orbital. The excitation \(1b_1 \rightarrow 4a_1\) produces a \(B_1\) excited state (\(b_1 \otimes a_1 = b_1\)), and the test asks whether \(B_1 \otimes \Gamma_\mu \otimes A_1\) can reach \(A_1\). It can — pick the \(x\) component, which is itself \(B_1\), and \(B_1 \otimes B_1 = A_1\). Allowed, and allowed specifically for light polarized along \(x\), perpendicular to the molecular plane: the character table hands you the polarization of the band as a free bonus, which is exactly the kind of information oriented-crystal and poled-film spectroscopy trades on.
Second, formaldehyde — the same molecule whose excited states an early post on this blog computed with four density functionals. The famous low-energy band is the \(n \rightarrow \pi^*\) excitation: the in-plane oxygen lone pair (\(b_2\)) into the out-of-plane \(\pi^*\) (\(b_1\)), giving an excited state of species \(b_2 \otimes b_1 = A_2\). Now look back at Table 1: no component of the dipole operator transforms as \(A_2\). There is no polarization of light — none — for which \(A_2 \otimes \Gamma_\mu \otimes A_1\) contains \(A_1\). The transition is symmetry-forbidden, full stop. That TD-DFT study found the \(n \rightarrow \pi^*\) transition near 4 eV with “negligible oscillator strength” in every functional — the computer rediscovering, numerically, a zero that group theory hands you on one line. The experimental band obliges: it shows up around 300 nm with a molar absorptivity of order ten, four orders of magnitude below a fully allowed transition1.
One more subtlety the parity argument alone would miss: a molecule can lack an inversion center and still forbid transitions, because any symmetry operation can drive the cancellation. Benzene has an inversion center and a six-fold axis and mirrors (\(D_{6h}\)), and its three famous ultraviolet bands are a ladder of exactly this effect — all three are \(g \rightarrow u\), parity-allowed, yet two of them are killed by the ring’s other symmetry elements. The transitions to \(^1B_{2u}\) (254 nm) and \(^1B_{1u}\) (204 nm) are orbitally forbidden in the full group; only \(^1E_{1u}\) (~180 nm) is allowed5,6.
The intensity ladder
Put numbers on the verdicts and the spectrum organizes itself into a ladder spanning six or seven orders of magnitude (Figure 3) — which is why “allowed or forbidden?” is usually the first question worth asking about any band, ahead of anything quantitative.
Figure 3. The intensity ladder: where symmetry verdicts land on a logarithmic absorptivity axis. A — benzene’s \(^1E_{1u}\) band near 180 nm, fully allowed, \(\varepsilon \sim 6 \times 10^4\); B — benzene’s \(^1B_{1u}\) band at 204 nm, orbitally forbidden but strongly vibronically assisted, \(\varepsilon \approx 8{,}000\); C — benzene’s \(^1B_{2u}\) band at 254 nm, orbitally forbidden, \(\varepsilon \approx 240\); D — tetrahedral \([\mathrm{CoCl_4}]^{2-}\), \(d\)–\(d\) with no inversion center to forbid it, \(\varepsilon \sim 10^2\); E — octahedral \([\mathrm{Co(H_2O)_6}]^{2+}\), Laporte-forbidden \(d\)–\(d\), \(\varepsilon \sim 5\); F — octahedral \([\mathrm{Mn(H_2O)_6}]^{2+}\), forbidden by parity and spin at once, \(\varepsilon \lesssim 10^{-1}\). Each rung down the ladder is another selection rule engaged; top to bottom spans roughly six orders of magnitude1,5.
The ladder’s bottom rung introduces the one selection rule this post has not yet named, because it is not spatial at all. The dipole operator does nothing to spin, so the spin state must be the same before and after: \(\Delta S = 0\), the spin selection rule. High-spin Mn²⁺ is a \(d^5\) ion with all five spins parallel, and every \(d\)–\(d\) excitation must flip one — so every visible transition of \([\mathrm{Mn(H_2O)_6}]^{2+}\) is spin-forbidden and Laporte-forbidden simultaneously, and manganese(II) solutions are nearly colorless, a hundred times paler than the merely Laporte-forbidden pink of cobalt hexahydrate. The same rule, run in emission, is why phosphorescence — emission from a triplet state back to a singlet ground state — is slow enough to watch glow-in-the-dark stars fade for minutes: the transition is spin-forbidden, its rate suppressed by orders of magnitude7.
Why forbidden bands appear anyway
Every forbidden band in Figure 3 has a nonzero \(\varepsilon\) — you can see viridian, after all, and benzene’s 254 nm band is the workhorse of every undergraduate UV lab. “Forbidden” evidently does not mean silent. It means the leading term vanishes: the integral is zero for a molecule frozen at its symmetric equilibrium geometry, interacting with light through the electric dipole term only, with spin decoupled from space. Each of those clauses is an approximation, and each has a leak.
The biggest leak is that molecules do not sit still. A vibrating molecule spends most of its time away from the symmetric geometry the selection rule was derived at; an asymmetric vibration transiently destroys the inversion center, and during that excursion the transition borrows a little intensity from allowed bands nearby. This is vibronic coupling, worked out by Herzberg and Teller in 19338, and it is the mechanism behind most of what you actually see in a “forbidden” band: the \(d\)–\(d\) colors of octahedral complexes and benzene’s 254 nm band alike are vibronically stolen intensity, which is why that benzene band appears not as one peak but as a comb of vibrational sub-bands riding the enabling vibration. The stolen goods are meager — a hundred here, a few thousand there, against the tens of thousands of a truly allowed band — which is exactly the spacing of the ladder’s rungs.
The other leaks are quieter. Spin–orbit coupling entangles spin with space and lets spin-forbidden transitions proceed feebly — fast in heavy atoms, glacial in light ones, which is why Mn²⁺ stays nearly colorless while iodine-containing phosphors glow efficiently. Static distortions do permanently what vibrations do transiently: a crystal site squeezed away from perfect octahedral symmetry is a little bit allowed all the time. And the electric dipole is only the first term of the light–matter interaction; magnetic-dipole and electric-quadrupole terms have their own, opposite selection rules (\(g \rightarrow g\) allowed!), just weaker by factors of \(10^5\) or so — visible mainly when the dipole channel is fully closed. The practical summary: forbidden transitions are weak, not absent, and how weak tells you which leak feeds them.
The same gate, one floor up
One more payoff, and it closes a loop with the other half of this blog’s science series. The parity argument of Figure 2 applies to any observable, not just transition dipoles. The first hyperpolarizability \(\beta\) — the molecular quantity the Pockels-effect post measured and the RLC post spent five sections trying to wire into a circuit — carries three spatial indices, so under inversion it flips sign three times: \(\beta \rightarrow -\beta\). In a centrosymmetric molecule, \(\beta\) must equal its own negative. Every centrosymmetric molecule has \(\beta = 0\) identically — the Laporte rule, one floor up. That is why every chromophore in the electro-optics literature is a donor–bridge–acceptor asymmetric by construction, and why a poled film loses its electro-optic response the moment its chromophores disorder back toward a centrosymmetric average: symmetry does not merely mute the response, it erases it. The dial from forbidden to allowed that the pigments post drew for absorption is the same dial the nonlinear-optics posts turn when they break a molecule’s symmetry on purpose.
So the pigment series’ promissory note cashes out to this: a spectrum has two independent axes, and symmetry owns one of them outright. The energy gap — set by conjugation length, ligand field, charge-transfer distance — decides where a band sits, and the whole pigment taxonomy is a catalogue of ways to place it. Whether the band is a whisper or a shout is a different question with a different owner: one integral, zero or not, adjudicated by the symmetry operations of the molecule in a table you can fit on an index card. Viridian is pale because an octahedron has a center; cobalt blue tints strongly because a tetrahedron doesn’t; YInMn was engineered by choosing a site where the gate stands open3,9. Forbidden or allowed is not a fact about what light wants to do — it is a fact about what a shape permits.
This post states selection rules in their leading-order electric-dipole form; the leak terms — vibronic, spin–orbit, multipolar — are treated qualitatively, and the representative absorptivities in Figure 3 are textbook order-of-magnitude values for aqueous complexes and vapor/solution benzene, not precision data. Any errors of translation are mine.